![]()

![]()
![]()
Band Structure via Quantum Espresso
Contents
- Prerequisites
- Overview
- 1. relax: Ionic relaxation
- 2. nscf: Performing a non-self-consistent field calculation
- 3. bands: Computing the band structure
- 4. plotbands: Plotting the bands
Prerequisites
Before getting started, you will want to make sure you have access to the following software:
- Quantum Espresso 5.2.0 (or greater)
- xcrysden 1.5.60 (other verisions likely work)
For installation instructions on these visit the software page.
Overview
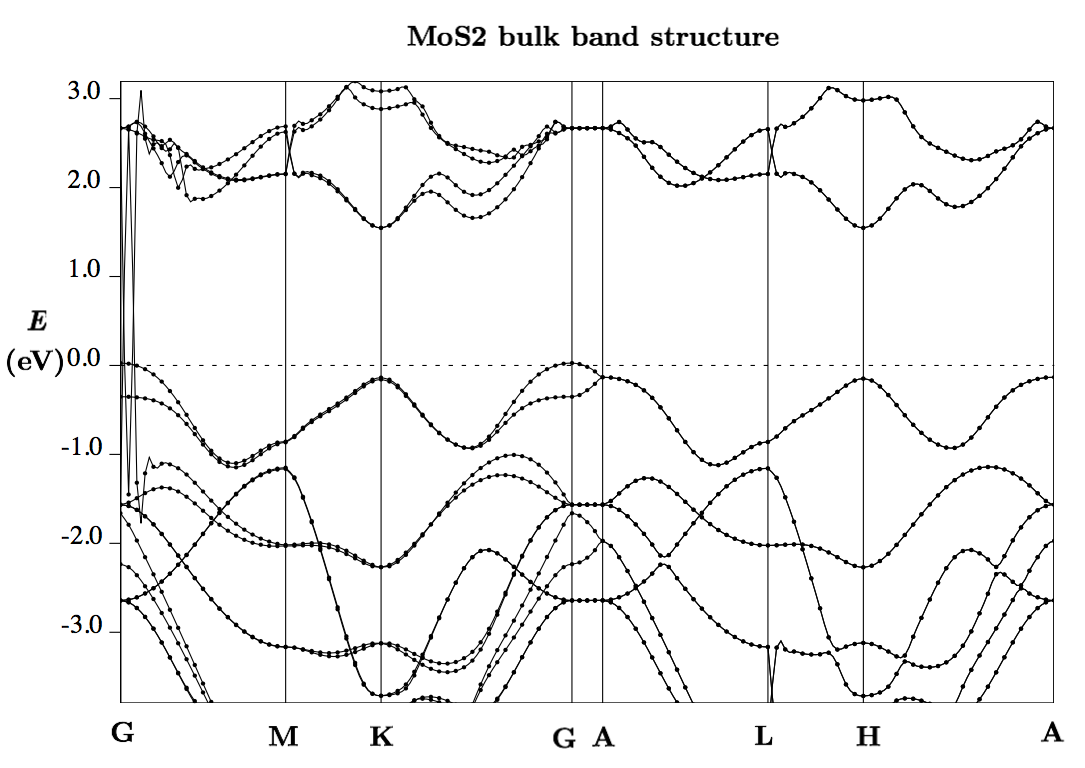
In this tutorial, we will calculate the band structure of bulk 2H-phase MoS$_2$. The bulk crystal is expected to be a semiconductor and have an indirect band gap. Indeed, we find both of these properties here using a PBE exchange-correlation potential. The results are very similar to the results in the VASP tutorial, which also used a PBE exchange-correlation potential.
Similar to VASP, a band structure calculation using Espresso involves a sequence of simulations. We can divide these into 4 parts:
- Relaxation of the atomic positions (uses
pw.x) - Non self-consistent (nscf) calculation (uses
pw.x) - Band calculation (uses
pw.x) - Plotting of the bands (uses
bands.xandplotband.x)
Since there are 4 parts, I prefer setting up a directory structure that corresponds to each of the calculuations. In the root directory, I have
$ ls
1-relax/ 2-nscf/ 3-bands/ 4-plotbands/ out/
Here, the numbered folders contain input files, while the out/
folder contains all of the Quantum Espresso-generated output, such as
wavefunctions, xml files, and everything else that is generated when
running Espresso. It is important to make sure that all input files
in the numbered subdirectories have the same prefix and outdir,
where outdir = '../out' inside each numbered folder. Below, we will
go over the calculations done in each subfolder in detail.
Files for this tutorial
All files can be found at this git repo. Alternatively, clone them via
$ git clone https://github.com/rehnd/MoS2-band-structure.git
General note about MPI
For the first 3 calculations, you should run with the same number of
processors for each of the calculations. The reason for this is that
Espresso saves wave functions and other data in files corresponding to
processor number. I have not experimented much with trying to change
the number of processors for subsequent calcualtions; I always run
with the same number of processors for steps 1-3, which all use pw.x.
However, step 4 (generating the bands files and plotting the bands)
can be done in serial. This is because part 4 uses bands.x and
plotband.x (not pw.x).
1. relax: Ionic relaxation
Input files for step 1: MoS2-2H.relax.pw.in
The first step is to relax the ion positions within the cell. This is
done using pw.x.
After running pw.x (which is set up in the run.sbatch script), you
will want to find the relaxed ion positions towards the end of the
output file.
Steps for running the relaxation:
$ cd 1-relax
$ sbatch run.sbatch
$ cat *.out ## --> look for the ATOMIC_POSITIONS near the bottom of the output file
Above, the .sbatch file contains:
mpirun -np X /path/to/espresso/bin/pw.x < input-file.in > output-file.out
You should notice a folder called out in the parent directory of 1-relax.
2. nscf: Performing a non-self-consistent field calculation
Input files for step 2: MoS2-2H.nscf.pw.in
After running the relaxation, you will want to copy the final ion
positions from the output of the relaxation and place them in an input
file in 2-nscf. This file should look like the following: (link
here)
There are 4 important differences between the input file from
1-relax and 2-nscf:
- Under
&control, you seecalculation = 'nscf', instead ofrelax. - The
ATOMIC_POSITIONSshould be updated in thenscfcalculation to be the ion positions after the relaxation. - The density of K_POINTS can (should) be higher in the
nscfcalculation - You can remove the
&ionstag in thenscfcalculation, because no ionic relaxation is needed. (If you forget to remove it, Espresso simply ignores the &ions tag by default)
3. bands: Computing the band structure
Input files for step 3: MoS2-2H.bands.pw.in
This step consists of 2 substeps. The first requirement is to choose a
list of k-points for the band structure. The easiest way to do this is
to use xcrysden, though you can use any method you like to do
so. The second step is to modify the input file and run the
calculation. Both steps will be discussed below.
a. Using xcrysden
First, you will want to install xcrysden. I have found that the
easiest way on Mac is to use MacPorts. More information is found
here.
The next step is to open a PW input file:
Once it is open, you should be able to rotate the unit cell around, as shown here:

To create a k-points list, go to the Tools tab:
Next, you need to select the k-space path that you want.
Useful: Check the “Display Reciprocal Vectors” box to see where the reciprocal space vectors are.
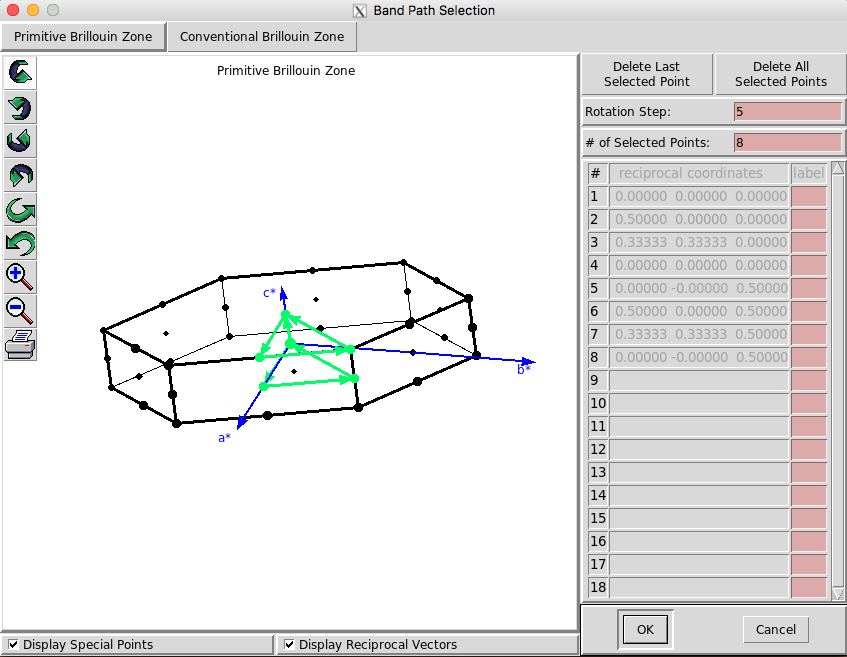
Next, seleck ‘Ok’ and choose the total number of k points along that path

Finally, save as a .pwscf file. You need to enter a filename that
ends with .pwscf to get it to work right.
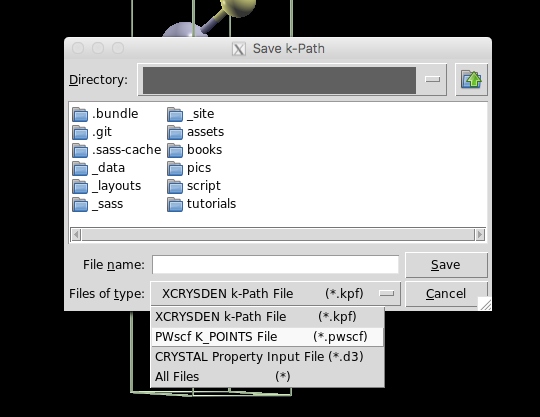
This will output a file starting with K_POINTS, a line with the
total number of k points in the file, and then a list of all k
points. This file needs to be appended to the input file you will use
to do the bands calculation, e.g.
$ cat output.pwscf >> inputfile.in
b. Modifying the input file and running
After generating a list of K_POINTS from xcrysden, you want to
copy that list to the K_POINTS section of the input file. (You need
to delete the previous K_POINTS list if you copied the input file
from step 1 or 2.
There are 3 important things to keep in mind about this file:
- The
K_POINTSsection of the file will be very different (and can be generated usingxcrysden) - You should continue to use the relaxed ion positions in
ATOMIC_POSITIONS - As with the
nscfcalculation, you do not need the&ionstag in the input file.
After adding the K_POINTS, run pw.x just like before on this new input file.
4. plotbands: Plotting the bands
This portion does not have to be done using MPI and should probably be run in serial. The input file for this is particularly simple, shown here:
&bands
prefix = "MoS2-2H"
outdir = "../out"
filband = "MoS2-2H.bands.dat" ! Name of the output file with data
/
To run this, you can use the provided run.sbatch script in the
4-plotbands directory, or simply run the following from the
commandline:
$ /path/to/espresso/bin/bands.x < input-file.in > output-file.out
After doing this, you will execute plotband.x, also located in
bin. Executing this will prompt you for 1) an input file, 2) an
energy range, 3) an output file(xmgr format) name, and 4) the output
file name for the postscript (.ps) plot generated.
For this example, I use
$ /path/to/espresso/bin/plotband.x
Input file > MoS2-2H.bands.dat # (same name specified by 'filband' above)
Reading xx bands at xxx k-points
Range: -8.360 8.9960 Emin, Emax > -8.3, 9.0
...
output file (xmgr) > bands.xmgr
...
output file (ps) > bands.ps
Efermi > 3.0 # you will have to choose this accordingly
deltaE, reference E > 1, 2 # deltaE = energy spacing on y-axis;
# reference E = zero of energy
After this, you should be able to open the .ps file and can rerun
plotband.x as necessary to get the desired look.
After playing with parameters and adding axes labels, you should end up with something like this: